| ABORTO SEPTICO
FISIOPATOLOGÍA
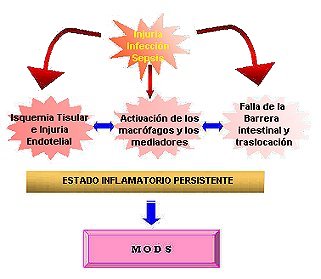
El grave cuadro de shock, en el que desencadena en algunas oportunidades el aborto séptico y donde son infructuosos todos los esfuerzos para tratarlo, pese a la celeridad y la intensidad de los tratamientos realizados (legrados, histerectomías, asistencia respiratoria mecánica, hemodiálisis precoz, uso de drogas vasoactivas, etc.), responde a la activación de una cantidad muy grande de mediadores: químicos, llamadas citokinas de las que se conocen más de 100;2.3.6.7 la activación de los polimorfonucleares, el fenómeno de la isquemia-reperfusión, que en realidad, aunque se los describe por separado (para una mejor comprensión), no es en realidad otra cosa que distintos mecanismos íntimamente relacionados unos con otros que se ponen en marcha en forma secuencial y se autoalimentan por mecanismos de feed-back. El fenómeno inicial, en la sepsis, que pone en marcha el complejo proceso de la cascada inflamatoria conjuntamente con la lesión panendotelial vascular está desencadenado por bacterias (Gram - y Gram +), más específicamente los componentes de la pared bacteriana (predominantemente en los Gram negativos), aunque también son capaces de desencadenar este fenómeno los hongos, parásitos y virus. Estos microorganismos, al producir infección, activan dos canales de respuesta: humoral y celular.15.16 Estas, en última instancia actúan sinérgicamente y se autoalimentan mediante complejos mecanismos de feed-back.10.17 Los avances, recientemente logrados, en biología molecular e ingeniería genética pusieron luz sobre algunos aspectos relacionados con la evolución de los cuadros sépticos. Está claro, ahora, que el fenómeno de estado inflamatorio sistémico, la aparición de shock séptico y la evolución al síndrome de disfunción multiorgánica (MODS), no sucede directamente en respuesta a factores exógenos, sino como consecuencia de la acción de mediadores producidos por el propio huésped.16 En la génesis del MODS, cuadro que fue descripto por primera vez por Tilney en 1973, en pacientes de Terapia Intensiva a raíz del sostén de la vida con los cuidados propios de estas áreas de atención médica que sigue al cuadro de sepsis, hoy se habla de tres mecanismos que actúan en forma simultánea (figura 2):
Aunque quizá exceda los propósitos de este escrito, vamos a describir brevemente los mediadores más conocidos que están involucrados en el desarrollo del shock séptico y el fallo múltiple de órganos. El mecanismo de los mediadores Este mecanismo involucra la estimulación de los macrófagos y éstos a su vez conducen a una sobreproducción de citokinas tales como interleukina 1 (IL-1), Factor de Necrosis Tumoral a (TNFa), IL-6 e IL-8. Estas citokinas activan la producción de mediadores secundarios que incluyen: óxido nitroso (NO) antiguamente conocido como factor de relajación endotelial, metabolitos del ácido araquidónico, bradikinina e histamina, los cuales activan a los neutrófilos y células endoteliales perpetuando la injuria tisular. Este mecanismo se perpetua a si mismo, produciendo lo que se conoce como efecto dominó.16.17.18 Experimentalmente se ha logrado inducir y reproducir la respuesta séptica por la inyección de agentes inflamatorios, endotoxinas, o citokinas como el TNF-a e IL-1. Dado que los mediadores inflamatorios tienen un efecto benéfico en situaciones normales, es difícil explicar porqué hablamos de su efecto dańino y por lo tanto determinar en cual situación clínica el bloqueo de la cascada de mediadores vs su estimulación puede ser beneficiosa. Haslett, ha esgrimido una hipótesis molecular. Sugiere que el síndrome séptico se debe al fallo de los mecanismos involucrados en la inflamación. Según esta hipótesis estaría alterado el proceso de apoptosis. Los neutrófilos en proceso apoptótico son reconocidos y fagocitados por los macrófagos sin liberación de proteasas ni radicales libres. Esto establece dos posibilidades: que falle el proceso de apoptosis de los neutrófilos y/o que haya inhabilidad de los macrófagos para reconocer o fagocitar a estos neutrófilos en proceso apoptótico.16.18 Este concepto encuentra sustento en la evidencia que las citokinas inflamatorias IL-1, IL-6, TNFa y las endotoxinas retardan la apoptosis de los neutrófilos. En la tabla 3 se muestran algunos de los mediadores más conocidos y sus efectos.
Figura 2 El mecanismo de la alteracion de la microcirculacion y el fenomeno de la isquemia/reperfusion Como todos sabemos, uno de los elementos diagnósticos para poder hablar de shock es, a grandes rasgos, la hipotensión arterial. Por este motivo se postula que la injuria orgánica esta relacionada a la isquemia y/o injuria endotelial vascular. Proviene en parte de la observación que la injuria (isquemia) seguida de un episodio de isquemia/reperfusión (como fenómeno segundo o second hit) puede conducir al desarrollo del MODS. Esta hipótesis microcirculatoria considera tres mecanismos:
La importancia de la isquemia esta dada en que el fracaso para mantener una adecuada disponibilidad y liberación de O2 a los tejidos. Los estudios de biología molecular establecieron que las células endoteliales son partícipes activos en la regulación del flujo sanguíneo, coagulación e inflamación, junto con los neutrófilos circulantes parecen ser los promotores de la isquemia y la injuria. Este paradigma, de interacción entre los leucocitos y el endotelio, que provoca injuria tisular, parece ser la vía patogénica común a diversos factores iniciales incluyendo bacterias, endotoxinas, citokinas e isquemia. Las células endoteliales una vez activadas, expresan receptores de superficie: ELAM-1 e ICAM-1 que promueven la adherencia y activación leucocitaria. Esto promueve la trombosis microcirculatoria y la injuria endotelial mediada por leucocitos. Así la activación de las células endoteliales resulta en isquemia tisular y en última instancia conduce al fallo orgánico, luego la posibilidad de la aparición del MODS. En el otro extremo, la inducción de citokinas es de indudable beneficio para el control y eliminación de patógenos bacterianos. Este hecho, el balance entre lo útil y lo dańino, que parece contradictorio, es válido y permanece aún sin una clara explicación. Pero, obviamente, hay una neta interacción entre la activación de los mediadores y el dańo endotelial mediado por la isquemia. La interacción entre neutrófilos/células endoteliales/receptores de superficie son necesarios para la erradicación bacteriana, y la intervención para lograr un efecto de down-regulation de este proceso puede conducir a efectos indeseables.16.17.18 Aparentemente, la reperfusión que sigue al fenómeno de isquemia puede ser el elemento más importante en la patogénesis del MODS que el período de isquemia mismo. Aunque la restauración del flujo es absolutamente necesaria para la sobrevida del órgano, puede inducir o exacerbar la extensión de la injuria isquémica a través de la generación de radicales libres, hecho este que puede ser prevenido por la administración de agentes que "limpien" (scavengers) o bloqueen la generación de radicales libres. Hay muchas fuentes biológicas de radicales libres, pero la mayor fuente parece ser la vía de la xantino-oxidasa y la de los leucocitos activados. Aunque no se ha probado, el hecho que la conversión de la xantino dehidrogenasa a xantino oxidasa toma solo 10 segundos en el intestino, 8 minutos en corazón y 30 minutos en hígado, rińón y pulmón, podría explicar la susceptibilidad diferente de estos órganos a la injuria tisular mediada por isquemia/reperfusión.
El concepto que el intestino es el motor del fallo múltiple de órganos fue propuesto por Meakins y Marshall. Permite, en parte explicar la observación clínica que el 30% de los pacientes con bacteriemia o sepsis que morían de MODS, no tenían foco infeccioso demostrable. Se pensó entonces, que el tubo digestivo con sus bacterias intraluminales podía ser el "culpable" de la producción de bacteriemias. Otro hecho importante es que algunas infecciones en pacientes críticos son causadas por gérmenes que normalmente son encontrados en la flora entérica. Este fenómeno conocido como traslocación bacteriana, se observó luego de injuria tal como: shock séptico y hemorrágico, trauma, quemaduras, malnutrición, el fenómeno de isquemia/reperfusión e inflamación, todas estas entidades que predisponen al desarrollo del MODS.16.18 La pérdida de la función de barrera, que ejerce el intestino, es un pre-requisito para la traslocación bacteriana. Hay varios hechos que alteran esta barrera uno de ellos el uso de antibióticos de amplio espectro que causa alteración de la ecología intestinal. El crecimiento excesivo de gérmenes Gram negativos o cándida predispone a traslocación bacteriana. El uso de nutrición parenteral compromete la inmunidad intestinal y las defensas mecánicas. A pesar de todo esto hay datos contradictorios. No se ha podido demostrar una correlación puntual entre la traslocación bacteriana y las complicaciones infecciosas. Una probable explicación habla que en realidad es la endotoxina antes que la traslocación bacteriana per se, el factor crítico que inicia y perpetua el MODS y por consiguiente el marcador de la evolución del padecimiento del paciente. Probablemente, el tubo digestivo es un componente más que genera factores múltiples y secuenciales que inician el MODS. El tubo digestivo puede considerarse como generador y mantenedor del proceso inflamatorio, magnificado y descontrolado tal como aparece en la disfunción multiorgánica. Recientemente quedó evidenciado que el tubo digestivo es productor de citokinas (por ej. IL-6, TNF-a, etc.) en respuesta al shock, aún en ausencia de traslocación bacteriana. El tubo digestivo, actuando en forma sinérgica con las células inmunes responden al shock con un mecanismo de up-regulation en la expresión de las citokinas y disminuyendo la respuesta inmune de la IgA a los antígenos intestinales. Dado que la Ig A es una inmunoglobulina no flogótica (no activa al Complemento) su reducción podría llegar a ser un factor que exacerbe la respuesta inflamatoria. El mecanismo intestinal se suma a la de los mediadores macrofágicos y al de la isquemia/reperfusión, que funcionaría como una fuente adicional de inflamación en el modelo golpes múltiples (hit-multiple) del MODS.16.17.18 Según este modelo, el desarrollo del MODS no estaría en relación a la gravedad de la injuria inicial, sino dependería de injurias sucesivas (hemorragias, shock, sepsis, stress quirúrgico o anestésico, etc.), esto a su vez generaría una secreción constante, aunque no elevadas, de las diversas citokinas y los demás componentes de la cascada inflamatoria. Tratando de unir secuencialmente los conceptos expresados, se podrían describir los sucesos evolutivos del shock séptico como siguen. Definido el foco infeccioso (absceso, celulitis, endocarditis, peritonitis, foco gineco-obstétrico en nuestro caso), la etapa siguiente sería la invasión al torrente sanguíneo de bacterias (bacteriemia) o sus productos (endo o exotoxinas). Una vez que se ha producido este suceso se activan las defensas del huésped: se pone en marcha el sistema mononuclear-macrofágico (monocitos, macrófagos, neutrófilos), se activan las células endoteliales. Estos en su conjunto ponen en marcha lo que se conoce con el nombre de pánico inmunológico al liberar mediadores de la cascada inflamatoria (TNF, IL-1, IL-2....IL-12, Complemento, trastornos en la Coagulación, Endorfinas, Eicosanoides, Radicales Libres, Interferón, Kininas, Oxido Nítrico). Este conjunto de mediadores inflamatorios tiene varias consecuencias fisiopatológicas, siendo quizá, las más importantes por los efectos hemodinámicos, las siguientes:
Ambos hechos desencadenan insuficiencia circulatoria expresada por acidosis láctica, alteraciones de la microcirculación (trombosis-isquemia-reperfusión), descenso de la resistencia vascular sistémica, aumento del consumo de O2, con lo que se conoce como dependencia patológica (que luego veremos). Esto produce tres eventos que interaccionan entre sí:
Por todo lo dicho anteriormente, es entendible que la evolución microbiológica sea independiente de la evolución clínica y que la mortalidad, por lo tanto no dependa del germen causal.17.18 |
||||||||||||||||||||||||||||||||||||||